
Welcome to the Raschke Group
New and distinct physical and chemical properties of matter emerge on the nanoscale when the structure size of the material becomes comparable to the mean-free path of the electrons or the scattering length-scale of phonons (finite-size effects). This can even occur in nominally homogeneous bulk materials in correlated materials (domain formation). The research in our group is centered around the development and unique applications of nano-optical spectroscopies that enable both nanometer spatial and femtosecond temporal resolution.
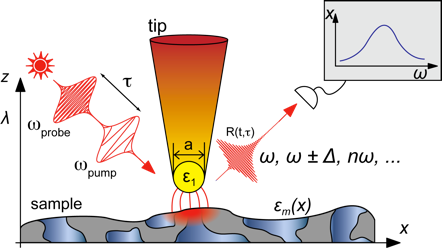

The optical antenna properties of metallic nanostructures allow one to concentrate and locally enhance optical fields to nanometer dimensions. This can be explored and applied for scanning probe optical near-field microscopy with nanometer spatial and femtosecond temporal resolution and sensitivity down to the single molecule level. Being compatible with a broad range of optical spectroscopies including time-resolved and nonlinear methods, we are making use of the technique for a broad range of applications. These inlcude:
- Study of the competing phases and nano-domain formation in transition metal oxides (multi-ferroic order, metal-insulator transition) and other quantum many body systems addressing the fundmental physics behind the rich behavior of these correlated electron systems.
- Ultrafast electron dynamics in metals and the control of coupling and nano-focusing of light using plasmonic and optical antenna structures.
- In situ investigations of supramolecular, biomolecular, and copolymer nanostructures, the nanomanipulation of optical molecular switches, and tuning the local optical coupling in molecular plasmonics.